
Public Citizen’s Health Research Group wants the FDA to deny speeded up approval of a new gene therapy treatment.
The advocacy group states that the treatment, SRP-9001 (Sarepta Therapeutics) for ambulatory Duchenne muscular dystrophy (DMD), intended for children aged four to seven, failed to show meaningful effectiveness in the only randomized study so far.
Public Citizen notes the treatment alerady has safety concerns — most notably that the viral vector needed to deliver this gene therapy cannot be used repeatedly, even for another treatment that might later prove to be safe and effective.
“If this potential treatment for DMD is fast-tracked with accelerated approval, other companies may be encouraged to rush unconfirmed gene therapies to the marketplace.”
Michael Abrams, M.P.H., Ph.D., senior health researcher at Public Citizen
Public Citizen’s petition to FDA adds to oral testimony given to FDA’s external advisory committee about SRP-9001 last month.
Public Citizen states that those advisory committee proceedings were wrongly “steeped in anecdotes over evidence,” and FDA officials at the meeting did not fully caution the committee about the potential consequences of accelerated approval.
“Once a therapy is approved by the FDA, via accelerated approval or otherwise, it is very difficult to remove it from the market. FDA officials should have been more forthcoming with the advisory committee about this ‘genie out of the bottle’ effect,” Abrams said.
A final FDA decision on the application is expected by Thursday (June 22).
Read the letter here or below.
Robert M. Califf, M.D.
Commissioner
Food and Drug Administration
U.S. Department of Health and Human Services
10903 New Hampshire Avenue
Silver Spring, MD 20993
Peter Marks, M.D., Ph.D.
Director
Center for Biologics Evaluation and Research
Food and Drug Administration
U.S. Department of Health and Human Services
10903 New Hampshire Avenue
Silver Spring, MD 20993
Re: SRP-9001(BLA # 12578/00, delandistrogene moxeparvovec, Sarepta Therapeutics, Inc.)
Dear Drs. Califf and Marks:
Public Citizen is a nonprofit consumer advocacy organization with over 500,000 members and supporters. We have no financial conflicts of interest regarding the development of treatments for Duchenne muscular dystrophy (DMD) or any other gene therapies.
In this letter, we urge the Food and Drug Administration (FDA) to reject the accelerated approval for SRP-9001, Biologics License Application (BLA) #12578/00, delandistrogene moxeparvovec, Sarepta Therapeutics, Inc. This biologic was the subject of the May 12, 2023, meeting of the Cellular, Tissue, and Gene Therapies Advisory Committee.[1] This letter supplements Public Citizen’s testimony presented during the open public hearing portion of that meeting.[2]
As stated in our testimony, we are deeply concerned about the efficacy and safety of SRP-9001 as a treatment for early-stage (ambulatory) DMD.
Our concerns are closely aligned with those expressed by the majority of the members of the advisory committee and the FDA’s scientific reviewers. Specifically, both the surrogate endpoint of in vivo muscle tissue micro-dystrophin levels and the safety of the gene therapy overall were called into question.[3] In this letter, we highlight two additional points.
I. Concerns about the evidentiary presentation and discussion at the advisory committee meeting
The discourse during the advisory committee hearing regarding clinical efficacy was too steeped in anecdotes over evidence. This concern is illustrated by the following exchange from a late portion of the meeting, immediately prior to the committee vote:[4]
Eric Crombez, M.D. (industry representative): “We’ve been talking a lot about these videos [of children with DMD stair-climbing, running, jumping, swimming, presumably in miraculous response to the treatment with SRP-9001] … are they representative or are they outliers?”
Craig McDonald, M.D. (consultant for the sponsor): “These videos are not outliers. They really represent profound benefit…profound durable effect[s] we just don’t see in muscular dystrophy.”
Although that testimony was compelling, it is inconsistent with what is known about the variable functional curves pertaining to children with DMD and with the primary endpoint results of the clinical trial pivotal to this BLA. DMD is distinctively heterogeneous with regard to disease trajectory over the main age range studied (4–7 years old), as demonstrated by the following “spaghetti” plot, which shows changes in motor abilities (y axis) by age (x axis) for a very large population of DMD patients ranging in age from 2–17 years (see Figure 1 from the FDA briefing document).[5] Accordingly, it is quite plausible that some of the young patients included in the SRP-9001 trial may have appeared to respond to treatment simply because their natural (untreated) disease trajectory fell on the less severe end of that spectrum.
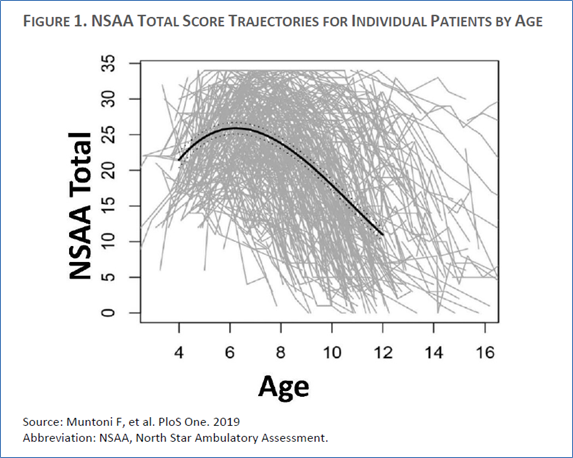
Additionally, and most importantly, if those videos truly represent non-outliers, i.e., relatively common patient responders to SRP-9001, then why does the single blinded, randomized trial (Study 102 Part 1, n = 41) not show such favorable effects on its primary endpoint (motor-functional performance) or via correlational analyses between that motor-functional outcome and the surrogate marker?[6] At present, the rational conclusion to be drawn from this single pivotal study is that either the treatment was not efficacious or it was far less efficacious than the isolated patient videos indicate. In the words of the FDA reviewers:
Therefore, despite demonstrating expression of Sarepta’s micro-dystrophin at Week 12 following infusion of SRP-9001, Study 102 Part 1 [the only randomized, double-blind, placebo-controlled trial completed to date] does not provide clear evidence that SRP-9001 is likely beneficial for ambulatory patients with DMD.[7]
II. Concerns about use of the accelerated approval pathway for SRP-9001
The potential fifth accelerated approval of a treatment for DMD should be placed in full context with the previous four treatments granted accelerated approval. Although they apply to distinctive mutational subpopulations, the prior treatments share the following important characteristics with SRP-9001:
- They all, in effect, aim to replace aberrant native dystrophin with a substitute molecule that is smaller and less complex than that endogenous protein.[8]Moreover, SRP-9001’s micro-dystrophin is by far the smallest of these truncated dystrophin replacements, just 138 kDa, whereas the original, native protein is 427 kDa.[9]
- Three of the four previous accelerated approvals were issued to the same company (Sarepta Therapeutics), which is also seeking the fifth accelerated approval.[10] Moreover, per recent reporting from STAT: “The company has generated distrust with the agency, because it failed to complete confirmatory trails for any of its three previous accelerated approvals, as required by FDA statute.”[11]
- None of the four exon-skipping treatments that previously received accelerated approval have been clinically verified through a required post-marketing study.[12]
During the May 12, 2023 meeting for SRP-9001, one advisory committee member (Donald B. Kohn, M.D.) asked the FDA to clarify what would happen if accelerated approval is granted for SRP-9001 and then the currently ongoing validating study does not show efficacy.[13] In response, one FDA official (Cecelia Witten, Ph.D., M.D.) said that the agency would review the emerging data and, if failed efficacy was confirmed, the agency would either work towards withdrawing SRP-9001 or they would see if the sponsor was willing to withdraw it, “which has sometimes happened.” Soon thereafter in the meeting, the Center for Biologics Evaluation and Research director (Peter Marks) stated that the 2022 user fee act passed by Congress added “some teeth” to the requirement that mandated post-marketing studies on accelerated approvals be conducted, and that a failed trial might trigger a future “advisory committee to have further discussions [about continuing approval].”[14] This exchange, however, did not convey to the agency’s external advisors that accelerated approval is often akin to prematurely “letting the genie out of the bottle.” That is, once the FDA approves a medication, it is very difficult to withdraw that drug from the market. Withdrawal of a drug, if it happens at all, invariably takes years.
Accordingly, we believe that FDA leaders were not sufficiently forthcoming about the implications of accelerated approval. It thus seems likely that the agency’s framing of this issue may have contributed to the 8-to-6 vote favoring approval.[15]
In our view, the advisory committee reached a distinctively mixed, but mostly negative, conclusion on SRP-9001, with more than half of the members concerned about the efficacy data and other members endorsing a potential treatment despite the concerns about efficacy, safety, and the regulatory history of very similar treatments for DMD.
Safety remains an important consideration, most notably the safety of any gene therapy that relies upon a viral vector.[16] In fact, though it has been dismissed as a rare adverse effect, a young man with DMD recently died because of a reaction to the adeno-associated viral vector (AAV).[17] The man apparently died from a rare reaction to the AAV, not rogue expression triggered by the CRISPR/Cas9 gene-editing complex deployed in this case. Moreover, related to this issue is the fact that use of AAV leads to an immune response that currently prohibits its future use. Thus, if an efficacious gene-editing therapy for DMD with an AAV is eventually developed, the patients who have already received another therapy, such as SRP-9001, will likely be unable to benefit.[18]
For the reasons discussed in this letter and by the advisory committee, the FDA should not grant accelerated approval of SRP-9001 for ambulatory DMD. Instead, the FDA should defer further regulatory consideration until after the ongoing confirmatory trial is completed. Although we are fully aware of the clinical situations of patients with ambulatory DMD, granting accelerated approval of SRP-9001 based on weak and unconvincing evidence is not a path forward.
Sincerely,
Michael T. Abrams, M.P.H., Ph.D.
Senior Health Researcher
Public Citizen’s Health Research Group
Robert Steinbrook, M.D.
Director
Public Citizen’s Health Research Group
[1] U.S. Food and Drug Administration. FDA briefing document, BLA 125781/00, drug name: delandistrogene moxeparvovec. Meeting of the Cellular, Tissue and Gene Therapies Advisory Committee. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed June 9, 2023. PDF p. 6, 27.
[2] Public Citizen. Testimony before the FDA’s meeting of the Cellular, Tissue and Gene Therapies Advisory Committee regarding inadequate evidence for accelerated approval for BLA# 12781/00 (SRP-9001; delandistrogene maxeparvovec) for the treatment of Duchenne Muscular Dystrophy. https://www.citizen.org/wp-content/uploads/2657.pdf. Accessed June 9, 2023.
[3] U.S. Food and Drug Administration. FDA briefing document, BLA 125781/00, drug name: delandistrogene moxeparvovec. Meeting of the Cellular, Tissue and Gene Therapies Advisory Committee. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed June 9, 2023. PDF p. 6.
[4] U.S. Food and Drug Administration. Recording of the 74th Cellular, Tissue, and Gene Advisory Committee. May 12, 2023. https://www.youtube.com/watch?v=k33d4h-CpGU. Accessed June 9, 2022. Time: 8:33:48 to 8:34:51.
[5] U.S. Food and Drug Administration. FDA briefing document, BLA 125781/00, drug name: delandistrogene moxeparvovec. Meeting of the Cellular, Tissue and Gene Therapies Advisory Committee. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed June 9, 2023. PDF p. 9.
[6] Ibid. PDF p. 6, 23-24, 32.
[7] Ibid. PDF p. 46.
[8] Patterson G, Conner H, Groneman, et al. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. Eur J Pharmacol. 2023;947(May 15):175675.
[9] U.S. Food and Drug Administration. FDA briefing document, BLA 125781/00, drug name: delandistrogene moxeparvovec. Meeting of the Cellular, Tissue and Gene Therapies Advisory Committee. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed June 9, 2023. PDF p. 14.
[10] Patterson G, Conner H, Groneman, et al. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. Eur J Pharmacol. 2023;947(May 15):175675.
[11] Mast J. What to know about a pivotal FDA hearing on Sarepta’s gene therapy for Duchene. STAT+ May 9, 2023.
[12] U.S. Food and Drug Administration. FDA briefing document, BLA 125781/00, drug name: delandistrogene moxeparvovec. Meeting of the Cellular, Tissue and Gene Therapies Advisory Committee. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed June 9, 2023. PDF p. 6.
[13] U.S. Food and Drug Administration. Recoding of the 74th Cellular, Tissue, and Gene Advisory Committee. May 12, 2023. https://www.youtube.com/watch?v=k33d4h-CpGU Accessed June 6, 2022. Time: 8:39:22 to 8:39:58.
[14] Ibid. Time: 8:50:02 to 8:51:53.
[15] Ibid. Time: 9:06:12.
[16] U.S. Food and Drug Administration. FDA briefing document, BLA 125781/00, drug name: delandistrogene moxeparvovec. Meeting of the Cellular, Tissue and Gene Therapies Advisory Committee. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed June 9, 2023. PDF p. 36.
[17] Mast J. Early findings in gene therapy death suggest CRISPR was not the cause. STAT+. April 26, 2023.
[18] U.S. Food and Drug Administration. FDA briefing document, BLA 125781/00, drug name: delandistrogene moxeparvovec. Meeting of the Cellular, Tissue and Gene Therapies Advisory Committee. May 12, 2023. https://www.fda.gov/media/168021/download. Accessed June 9, 2023. PDF p. 10.

Visit the Lemonade Mermaid Store today
Original and Custom items for Land or Sea Mermaids


